

How one family’s journey through the realm of rare disease led them to the newest frontier of precision genetic medicine.
BY TREY POPP | Photography by Jonathan Robert Willis and Glenda McCoy

David Faughn W’90 entered parenthood the way many fathers do: suddenly, hopefully, and at a bit of a loss about what to do. Katherine lay on her mother’s belly having spent 40 weeks within it. Her father was a foreigner to her. Or that’s how it seemed to Dave. The “ten months of uselessness” he had felt during Glenda’s pregnancy had not ended in the delivery room, he thought, just taken on a new form. He was as helpless as any new father. Helpless to feed her, helplessly unfamiliar to her, helplessly in love with her, already.
The family’s swift release from the hospital, bearing as it did the shock of responsibility, intensified the sensation.
“At first, we were strangers looking at each other,” he later recalled in a blog post: “me trying to figure out what to do, and she trying to figure out where mommy went.” But a bond was forming. Its profundity hit him one day on diaper duty. Katherine lay on the changing table while Dave made goofy faces, fishing for a smile. She unleashed a raucous belly laugh, a laugh as full-throated as Glenda’s. Dave “literally jumped in the air” and called Glenda in a rush of excitement. Her bemused response didn’t dent his elation. “At that moment, I became ‘daddy,’” he wrote, “and to the most wonderful girl who has ever been or ever will be.”
c.166G>A
The first year of Katherine’s life suggested the sort of alumni-note announcement that her father wasn’t actually the sort of person to write: “Katherine Belle Faughn was born on July 9, 2011, and is already well on her way to joining the Quaker Class of 2033.” Dave was an attorney for a Lexington, Kentucky, firm specializing in civil litigation and equine law. Glenda (who kept her birth name, McCoy) worked in the governor’s office as a project manager for the Kentucky Commission on Women. Katherine seemed hell-bent on beating her daycare classmates to developmental milestones. The nursery school staff nicknamed her “Flash” for how quickly she got to crawling—six months—and how speedy she was once she’d mastered it. Glenda began to fret that she’d have a nine-month-old walker on her hands, as though life weren’t hectic enough already.
But as her peers began catching up, Katherine seemed to stall. Dave and Glenda weren’t inclined to make a big deal out of it. They weren’t tiger parents. Dave’s aunt hadn’t walked until she was nearly two. She’d turned out fine. But the longer the delay persisted, the more it ate at Glenda.
“I would see so-and-so’s kid stand up at my feet and start walking across the room,” she recalls, “and Katherine would still crawl at everyone’s feet. And at 13 months she was still doing it. Everybody in her classroom was up and running around and I sat there thinking, why, why is my child not up and walking with them?”
c.815-27T>A
By 18 months, Katherine was in physical therapy. She seemed to possess the strength to walk, but whenever she tried her limbs would jerk, or her knees would lock, or she would take exaggerated strides, and she would tumble to the floor. She fell a lot—even when crawling, or simply sitting still. The physical therapist was stumped. An occupational therapist wondered if she suffered from a sensory-processing disorder. But then some of those issues seemed to improve. Meanwhile, Katherine kept being Katherine. She was a blond-haired, blue-eyed pixie with energy that wouldn’t quit. She liked hide-and-seek and dancing with her daddy. She commanded a vocabulary that was advanced for her age—yet her speech had begun to sound “thick” to Dave and Glenda. The confusion persisted.
Still unable to walk at her two-year pediatric check-up, Katherine was referred to the Cincinnati Children’s Hospital’s neurology department, which is reputed to be one of the best in the country.
On August 30, 2013, Katherine underwent a brain MRI. A doctor phoned Dave late in the day to discuss the results. Through a thicket of difficult terminology—lactate peaks, T2/FLAIR hyperintensities—one finding landed like a sucker punch: Katherine’s cerebellum was abnormally small.
c.693+1G>A
The cerebellum is a critical structure in the brain. Every creature with a backbone has a cerebellum. In humans, as in other animals, it is most strongly associated with motor control and coordination. So that solved the mystery of why Katherine had such a hard time keeping her balance. The underlying cause remained unknown, but the test results and the timing of Katherine’s symptoms strongly suggested a metabolic disorder. A team of CCH neurologists, metabolic specialists, and radiologists were confident they knew what it was: infantile neuroaxonal dystrophy, or INAD.
Dave had never heard of it before. INAD is caused by a rare genetic mutation inherited from both parents. Its frequency is unknown, but the disease is estimated to afflict fewer than 1 out of 200,000 newborns. An American is roughly 15 times likelier to be struck by lightning than by INAD. Anyone forced to choose would opt for lightning. By the time the doctor had finished talking, Dave understood two things. Katherine was going to die young. But first she would likely lose her ability to move, speak, swallow, see, and hear.
INAD progressively entraps children inside failing bodies. In time, Katherine’s skin might even lose the capacity to register the sensation of her parents’ touch.
c.311T>C
There was no cure— not even any experimental treatments in the pipeline.
Dave asked the most abject question a parent can utter: “Doctor, is there any hope?”
There was a pained and awkward pause. “You have a beautiful daughter,” the doctor replied. “You need to spend as much time with her as you can.”
The phrase “before she dies” was left unsaid, but that’s what the advice added up to. The clock was ticking.
p.L104P
Dave told Glenda. Her knees buckled. She crumpled to the floor, screaming. A period of time elapsed; pain voided her memory of it. Her eyes fell upon Katherine. Who was smiling but hungry. Who needed a fresh diaper. Who wanted a story before bedtime.
c.815-27T>C
“We took turns being strong for her,” says Dave. “We didn’t want her to miss out on anything, so we’d laugh and play with her. It’s like you’re putting on a face while the other person went out of the room and just curled up into a ball and died, or felt like they were dying. And then we’d just switch … put everything aside and just live in the moment with her.”
So life became a series of weighty moments and heartbroken retreats.
“You really do start to appreciate every single moment,” Dave says. “You’re not looking toward tomorrow, because tomorrow’s a nightmare.”
Glenda felt like they were hurtling through the stages of grief. “The sadness and the bargaining and the anger were all happening at once,” she recalls. “It just knocked us both down where we were pretty much in a fetal position,” wondering when they would reach the serenity of acceptance.
c.815-27T>C
Dave resisted. He typed “INAD” into his web browser and followed every link. He read blogs by families who’d gone through it, watched videos of afflicted children. What he found was not for the fainthearted. “Most of the things you stumble across on that disease are In Memoriam websites about children who died at about seven years old,” he says. “So it’s terrifying.”
Using Google Scholar, he combed the medical literature for INAD references. He researched every finding from Katherine’s MRI report, and began a list of anything besides INAD that might explain it. He investigated things that hadn’t turned up on the MRI, and became fixated on iron accumulation in the basal ganglia. It is a hallmark of INAD, but hadn’t been apparent in Katherine’s scan.
By his reckoning, there were four alternative explanations for Katherine’s condition. Each one was even rarer than INAD, but he became convinced that it had to be one of them.
“We were aware of what was happening to us psychologically,” Glenda remembers. “At that point you’re just grasping … so you wonder, ‘Are we just in the denial part of grief?’”
The medical team at Cincinnati Children’s Hospital listened politely to Dave’s thoughts. And remained confident in their diagnosis. Iron accumulation takes time. The disease may simply not yet have progressed that far in Katherine. She had INAD; they were over 90 percent sure of it.
Dave requested a genetic test, to know definitively. The family’s insurance carrier issued a denial of payment.
It was a $2,500 test to assess a single gene. If it yielded the desired result—negative for a mutation of the PLA2G6 gene—it could open the door to tests costing an order of magnitude more. Glenda had already left her job to care for Katherine full time. They opened their wallets, and started the process of selling their house.
c.166G>A
But for the fact she was still on all fours, Katherine was in many ways a typical two-year-old. She roamed the patio with a plastic watering can, pulling herself up on flowerpot edges to get at the dirt. She threw tea parties for a congregation of stuffed bunnies, all named Bibi—Bibi, New Bibi, Itty Bibi, Other Itty Bibi, and so on. She liked ordering her daddy to “sit” so she could fasten bows to his hair.
And when the test for the dreaded PLA2G6 gene mutation came back, she didn’t have it.
Yet the doctors still thought she had INAD—just an atypical case of it.
So far, Glenda hadn’t had the stomach to look at Dave’s web research. But now she wanted to see pictures and videos of children with INAD. And when she did, she couldn’t shake the same feeling Dave had: they didn’t look like Katherine. Even apart from the feeding tubes, “they had more weight on them, the look in their eyes was different,” she recalls.
But how do you ask a doctor to pay less attention to an MRI and more to the look in a little girl’s eyes?
Dave had come to believe Katherine suffered from a mitochondrial disease and decided to find a new doctor who specialized in them. The one they chose, at the Cleveland Clinic, was upfront about his opinion: Katherine did not present like a patient with mitochondrial disease. For one thing, she buzzed with energy. Mitochondria organelles are the cell’s energy centers; mito disease patients tend to lack strength and tire easily. And it just looked an awful lot like INAD.
“But is there hope?” Dave asked, again.
c.815-27T>C
Hope springs eternal in modern medicine. But some kinds of medical knowledge can extinguish it with brutal finality.
What is known about many rare genetic diseases is especially cruel. The protein mutation that causes fatal familial insomnia has been found in only a few dozen families worldwide, but it invariably afflicts its sufferers with sleepless delirium that ends in death. Children with Progeria syndrome, caused by a mutated LMNA gene, age prematurely and almost always die before 20. There are perhaps only 800 people in the world who carry a particular mutation in their ACVR1 gene, but they all suffer the same fate: an errant cellular-repair mechanism that converts their muscles and other soft tissues into bone, until their bodies essentially turn into statues.
There is no known way to reverse any of those diseases. And an INAD diagnosis is just as dire. Dave and Glenda wanted hope for Katherine. But what they really wanted from a doctor was a capacity for doubt.
c.667_668insCCTTGTGCTG
Sumit Parikh, who directs the Cleveland Clinic’s Neurogenetics, Metabolic & Mitochondrial Disease program, had an appreciation for the limits of diagnostic certainty. Even if Katherine had INAD, he told Dave and Glenda, the fact that it hadn’t been caused by the typical mutation raised the possibility that it might progress differently as well.
Parikh ordered new tests. As, one by one, each test failed to confirm INAD, the family journeyed deeper into a realm of more profound doubt. It held a feeling of deliverance—even if only provisional deliverance—for virtually any diagnosis carried a better prognosis than the one hanging over them. But chipping away at one hypothesis was bringing them no closer to an alternative.

“When you’re undiagnosed, you feel like a refugee,” Dave told me in October, using the same word Glenda had used at the time in a blog entry. “We all want a home, even if that home is the worst one imaginable,” she had written. “Any home is better than nothing.”
With all the specific tests and assays exhausted, one option remained: whole-exome sequencing. The exome consists of the one or two percent of the genome that codes for proteins. Sequencing it is therefore cheaper than sequencing the entire genome. But it is not cheap.
c.166G>A
As lawyers go, Dave is conflict-averse. “My job is to push up against things,” he says, “but it goes against my grain. I’m normally not a fighter.”
But when Humana Incorporated denied payment for Katherine’s first genetic test, he threw all his energy into overturning the decision. In a scathing 19-page appeal, he charged the company with misrepresenting the policy contract’s language in a bad-faith “decision to remain intentionally ignorant at the known cost of Katherine’s life.”
Humana coughed up a reimbursement.
Now, after obtaining a pre-approval for whole-exome sequencing, Dave and Glenda sent Katherine’s blood to be tested, along with their own. It was October of 2014. It would take around four months to get results.
c.815-27T>C
Almost immediately, Katherine pitched into a steep decline. She became clumsy. Her energy vanished. “I can’t climb anymore,” she would tell her parents. “Pick me up.” She had what looked like a seizure, with jerky movements, dry-heave vomiting, and a frightening lapse into non-responsiveness.
Emergency room physicians placed her on Keppra, a seizure medication, and eventually released her. Then the whole thing happened again.
In the run-up to Halloween, Katherine had been scrambling up hay bales and frolicking in piles of fallen leaves. Two weeks after Thanksgiving she seemed like a different child.
“For the first time, I believed, truly believed, that Katherine had INAD,” Dave wrote. “I had just been in denial. My daughter was dying in front of me. By Christmas, I was convinced that it would be her last.”
c.313G>T
Dave’s emotional strain spilled into public view. He devoted a blog entry to the suicide of Robin Williams, whose autopsy had revealed Lewy Body Dementia, another progressive neurological disorder with symptoms similar to INAD. His mother asked him if he was okay. He blogged about that. “No, I am not ok. I do not know that I will ever be ok again,” he wrote. “None of you want to read that. I am sorry to write it. Yet, that’s the truth.”
He confessed to having panic attacks, haunted by the thought that Katherine would lose her ability to speak while he was at work, and that he would get home too late to hear her say the word daddy again.
He also described a happiness of almost excruciating intensity.
“Laughter and joy are Katherine’s currency. She spends them freely. I am more alive than I have ever been. I feel more deeply than I’ve ever felt,” he wrote. “I see genuine goodness in people around me, in friends, family and complete strangers. People who reach out to lift our spirits and to help us practically and emotionally. I see my daughter in all children and love them for it.”
He remembers those three months as the worst period of his life.
c.815-27T>A
In January, Dave and Glenda took Katherine to Disney World, hoping she would be able to enjoy it. She was “wish-eligible” for assistance from the Make-A-Wish Foundation, but the family feared the application process would take more time than Katherine had, and weren’t sure her energy would be equal to the Magic Kingdom anyway. Upon arriving in Florida they were dealt two insults in quick succession. The first was a notice from Humana denying payment for the pre-approved whole-exome sequencing, which cost $27,000. The second was a notice from the hospital informing them that Katherine actually had no insurance at all and owed $55,000 in bills.
But it was a happy new year. Dave successfully appealed Humana’s denial and traced the hospital charges to a bureaucratic error that was resolved. More importantly, Katherine bounced back. She made it through the Disney marathon with energy to spare—holding her dad’s hands to jump up and down in delight under the nightly fireworks show—and soon was again her gleeful, scrambling self.
February brought the whole-exome results. They were definitive, and wiped the diagnostic slate entirely clean: Katherine had a mutation to a recessive nuclear gene called NUBPL.
Little was known about it. But in 2010 a mutation to that gene had been implicated in a rare mitochondrial Complex I disorder. Very rare: it had since been reported in just five families worldwide.
c.693+1G>A
Dave and Glenda wasted no time trying to figure out what that meant and what to do about it.
Before pulling the trigger on whole-exome sequencing, they had read a New Yorker article about Matt and Cristina Might, who in 2007 had a son with an affliction that stubbornly eluded diagnosis until whole-exome sequencing revealed a mutated gene called NGLY1. The only way to know if it explained the condition was to identify another patient with the same mutation and the same symptoms—but none was known. Matt, a computer scientist, posted an essay on the web that went viral. It was titled “Hunting Down My Son’s Killer” and contained detailed information about his son’s symptoms, lab results, and NGLY1. He envisioned it as a “reverse drag net”; if other families were experiencing the same thing and Googled their way to his post, together they might solve the mystery. Then they could band together and seek possible treatments.
“I didn’t sleep for three days after I read it,” Glenda says. They had initially started their blog, “Hope for Katherine Belle,” as a way to provide friends and family members with updates that were too painful to talk about in person. Suddenly it was clear that it could be more.
“I realized for the first time that we had a really big role to play in this,” Glenda says. Dave and Glenda published the genetic codes of Katherine’s mutation and those of other reported cases. (They are reproduced in the section breaks of this article.) They presented Katherine’s clinical progression in depth. And Glenda scoured the web and social media, reasoning that other families might have done some of the same things.
c.815-27T>C
In March 2015, they posted the first results of their search. “We want to introduce you to the Spooner Family and their daughters Cali and Ryann, both of whom have mutated NUBPL genes like Katherine,” they wrote. “We were undiagnosed for only two years … their oldest daughter was undiagnosed for thirteen years.”
The girls were six and 15 years old. Glenda had stumbled across a Facebook link to a documentary film about them. Watching it was a transformative experience for a mother consumed by fears that each birthday would be the last, and questions about what clothing her daughter would be buried in.
“That first glimpse at Cali Spooner’s face added years to my child’s life,” Glenda later recalled. “In her photograph I saw Katherine smiling back at the camera. For the first time, I saw Katherine as a teenager.” Meanwhile, six-year-old Ryann could walk independently.
c.579A>C
Identifying a community —even a vanishingly small one—catalyzed the Faughn family’s search for treatment options. They enrolled Katherine in a clinical trial of EPI-743, a potent antioxidant that targets a particular enzyme involved in cellular energy metabolism. They got her onto what is commonly called the “mito cocktail,” a blend of vitamins and supplements and antioxidants thought to reduce the excessive levels of free radicals resulting from mitochondrial dysfunction.
Predictably, Humana refused to cover the mito cocktail. (Vitamins and supplements are not considered medicines, nor is their manufacture subject to FDA oversight, so insurers commonly exclude them.) After a second insurer also declined to cover it, Dave took his crusade to the Kentucky legislature.
An attorney he had clerked for in law school, Sannie Overly, had become an influential state representative and a follower of Katherine’s blog. Dave wrote model legislation that would require Kentucky health insurers to cover mito cocktail formulations, and she sponsored it as an amendment to an existing bill. When it passed the Democratic-controlled House only to be voted down in the Republican-controlled Senate, Dave and Glenda targeted the Senate Majority Leader’s open Facebook page.
“We started posting videos of Katherine on there, saying, ‘How are you killing this bill that’s helping my daughter?’” Dave recalls. “And to his credit, he contacted us personally … We educated him on what the bill was for, and he came over to our side.”
The revived bill “came out of the committee at 15 till midnight on the last day of the session,” Dave recalls. “It was put in front of the House to vote on, and the Senate to vote on. It got signed at five minutes till midnight. And Sannie literally had one of her staffers run it from the Senate chambers into the governor’s office and got it there as the door was closing.”
The governor didn’t sign it, but didn’t veto it either, so Kentucky became the first state in the union to legislatively mandate that insurers cover mito cocktails.
c.166G>A
Mitochondrial disease exemplifies one of the thorniest challenges presented by rare diseases. In the United States, a disease is classified as “rare” when it affects fewer than 200,000 Americans. The vast majority of these are genetic in origin and affect a far tinier fraction. Such small patient populations severely limit the impetus for research and drug development. Yet roughly 7,000 rare diseases have been identified, and the National Institutes of Health estimate that collectively they affect 25–30 million Americans—more than cancer. The FDA has approved treatments for only about 5 percent of rare diseases.
An estimated 80,000 Americans suffer from mitochondrial disease—but mitochondrial dysfunction itself comes in so many flavors that a therapy that benefits one patient might be useless, or even harmful, for another.
The first mitochondrial genetic mutation causing human disease was identified 30 years ago by Douglas Wallace, who is now a professor of pathology and laboratory medicine at the Perelman School of Medicine, and director of the Center for Mitochondrial and Epigenomic Medicine at the Children’s Hospital of Philadelphia (CHOP).
“Now we know of nearly 400 different mutations in the mitochondrial DNA that can cause disease,” says Marni Falk, an associate professor of pediatrics at the Perelman School and executive director of CHOP’s Mitochondrial Medicine Frontier Program. Animals inherit mitochondria exclusively from their mothers. But the processes by which those organelles produce energy also involve nuclear DNA, which is inherited from both parents. And, says Falk, “we now know of 300 nuclear genes that can cause mitochondrial disease.”
The challenge presented by all that complexity is at the center of Falk’s research—which was about to expand to a new patient: Katherine Faughn.
c815-27T>C
As a medical resident in pediatrics and medical genetics in the early 2000s, Falk sometimes saw children with mitochondrial dysfunction at Case Western Reserve’s Rainbow Babies and Children’s Hospital in Cleveland.
“Nobody knew what to do,” she recalls. “The family would say, ‘What do we do about it?,’ and [the doctors] would say nothing. And they’d just stay in the ICU until they died. And it could be months.”
The geneticists couldn’t even counsel the parents about the odds another child would suffer the same fate: “We had no idea.”
Around the same time, one of her mentors, an anesthesiologist named Phil Morgan, happened to be part of a team investigating a common class of anesthetics. Volatile anesthetics (also known as inhalation anesthetics, like nitrous oxide) have the remarkable property of immobilizing a wide variety of animals—from humans to fruit flies to nematodes—at very similar concentrations. But their full mechanism of action remains unknown to the present day.
Looking at nematodes, the investigators found something interesting: a single genetic mutation dramatically increased worms’ sensitivity to volatile anesthetics. Morgan remarked that kids with mito disease were also often unusually sensitive. So the team drilled down into the nitty gritty of the gene—dubbed gas-1 for “general anesthetic sensitive”—and found that it was involved in the manufacture of mitochondrial Complex I.
Excited by the possibility of creating a genetic model that might finally answer the questions of mito disease families, Falk joined the search for other genes involved in Complex I.
240-kb deletion
The mitochondrial respiratory chain converts oxygen and simple sugars into molecules of chemical energy called ATP. The process involves five complexes, which can be envisioned as successive factories. Each factory feeds its product into the next one, while shunting byproducts toward other important cellular processes. Complex I is a “monster,” Falk says. It employs 45 subunits to create an enzyme, NADH, whose reduced form is also needed for 500 other reactions in the cell.
NADH is like a “big Lego structure,” Falk says, and the NUBPL gene helps some of the Lego blocks snap together.
Consider what that means. A mito patient who has trouble manufacturing a particular block might respond to a therapy that’s of no use at all to Katherine, whose difficulty lies not in making blocks but in connecting them. Similarly, if stray blocks are tumbling around Katherine’s cells, they could be causing types of trouble utterly foreign to a third mito patient. In a sense, each of those “mito disease” patients actually has a separate disease.
“Sometimes families call me,” Falk says, “and they say, ‘I read this. I read that. Which drug should I be on?’ And I say, ‘I don’t know. Nobody’s ever known.’”
So over the last couple years, she has built a research platform that aims, with an unprecedented level of precision, to find out.
On a sunny day in mid-October, one of her collaborators showed me how.
137-kb duplication
Christoph Seiler is a mutant zebrafish farmer. He breeds and analyzes genetically altered versions of the finger-sized fish, which are a well-established model organism in genetics research. As director of CHOP’s three-year-old zebrafish core facility, he oversees the technical wizardry at the heart of Falk’s research. Using a cutting edge gene-editing tool called CRISPR/Cas9, Seiler has been able to essentially insert the precise genetic mutations of Falk’s patients into zebrafish genomes, creating organisms that enable Falk to assess the resulting functional differences with the ultimate degree of specificity.
Falk uses other tools to make similar alterations to nematodes, which, like zebrafish, are champion reproducers with fast life cycles. (A robotic system dubbed the WorMotel, developed by Christopher Fang-Yen, the Wilf Family Term Assistant Professor of Bioengineering, permits automated analysis of tens of thousands of nematodes at a time—a dramatic leap from the manual observations long performed by sore-eyeballed graduate students.) She also edits patients’ cells, in vitro, to replace mutant genes with normal ones. Since the cells are identical in all other ways, any variations in their function can be attributed directly to the gene in question.
“We have about half a dozen different projects in the lab now that are based upon individual children with specific rare diseases in different parts of this process,” Falk says: “the electron transport chain Complex I subunit; the assembly of Complex IV; in this project, the assembly of Complex I; in another, the function of Complex V.”
In his lab in the Abramson Research Center, Seiler pulled out a petri dish swarming with a dozen or so zebrafish larvae. They were five days old, each about the size of a staple snipped in half. As offspring of adults possessing mutations to recessive genes, about a quarter of the juveniles had inherited both mutated copies. Seiler squirted a few drops of tricaine into their water, which quickly and temporarily put them to sleep. Using delicate tweezers, he lined up three fish in the focal field of a microscope.
Through their transparent skin, a difference was immediately clear. Two of the fish looked normal—having inherited at least one normal copy of the gene. But the third had an obviously malformed and shrunken air sac, and appeared to have liver abnormalities as well.
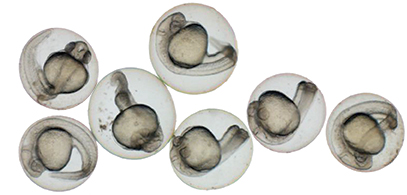
In October, Falk and Seiler were just beginning to try to create organisms possessing Katherine’s mutation. They might show anatomical differences, or might not.
“Mito disease kids often look normal,” Falk notes. “When the batteries don’t work in a doll, it’s not like the arm falls off, right? It’s just simply that they don’t walk, and they don’t talk.” Functional problems are more common than structural ones.
That’s another advantage zebrafish offer. Seiler flicked the rim of another petri dish and the fry scattered. “You can turn a light on and off to startle them, too,” he said. “They have this fast scooting motion with three turns of the tail. And you can study that and see if something’s abnormal. You can analyze movement very simply, or look at the trajectory of the swimming and see if that changes.” Or you can make zebrafish swim against a current and measure their endurance and recovery.
With anatomical and behavioral baselines established, Falk can then test existing drugs on the model organisms and see if anything has a positive impact. “We’re not interested in discovering some new drug,” Falk says. “We’re interested in an FDA drug, maybe one that exists for some other disease that I could just study in these kids, without having to spend 10 or 20 years developing.” That said, there’s no reason to rule out screens of novel drug candidates.
Some of the drugs she’s interested in don’t even target mitochondria at all. “There’s a lot of changes that happen outside the mitochondria in the cell when the mitochondria aren’t functioning. Maybe they’re part of the problem—those other changes,” she says. “If we could slow down the cell’s desire to fix everything by making more and more proteins, we can make the cell a lot healthier—and the organisms and the animals.”
“My dream for all this is that ultimately we’ll have lots of therapies that work,” she says. “But they might be different, or in different combinations, in different people.”
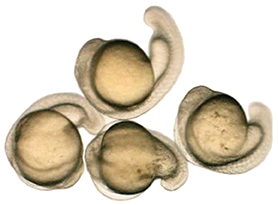
Successes with tiny groups of patients— even single patients—could in time have ramifications for far larger groups.
“Complex I not functioning properly is known to happen in Parkinson’s disease,” Falk notes. “And the dopaminergic neurons in Alzheimer’s disease. It happens in ALS. In diabetes. Things like heart attack or stroke, the parts of the brain that are damaged can wind up absolutely having a Complex I defect.
“If we can improve health in kids where the program is abnormal, then maybe we could also improve health” in a broader array of diseases related to aging. “And this is where Big Pharma gets interested.”
Orphan therapies, as treatments for some rare diseases are known, “often serve as platform technologies for future expansion across indications,” says Brian Corvino WG’11, a managing partner at the healthcare consultancy Decisions Resources Group. “If you look at just the past three years alone, 40 percent of all of the FDA approvals had an orphan drug designation,” he adds.
Sometimes drugs developed for a rare disease later find application in a more common one. Sometimes it goes in the other direction.
“Eight out of the top-10-selling drugs in the US in 2016 had some form of orphan drug designation at some point in its development,” Corvino says. “Now, that doesn’t represent a majority of the sales—but the orphan drug label collectively is an important one.”

Even the gene whose mutation makes Katherine seem like a one-in-a-billion rarity also illustrates how tightly all humanity is bound by our common genetic heritage. This past June, researchers at Harbin Medical University in China discovered that NUBPL plays a “vital role” in the metastasis of colorectal cancer. Modulation of the gene’s productivity, they proposed, could provide a novel therapeutic target for the second-leading cause of cancer death worldwide.

The creation of a Katherine Belle zebrafish will depend on laboratory luck and philanthropic generosity. The National Institute of General Medical Sciences supports Falk’s research, as does the CHOP Research Institute. She also receives pharmaceutical company funding. But philanthropy is part of the mix.
“Families go out and raise money, whether through silent auctions or through lemonade sales,” she says. “The money goes for the salaries and the reagents to actually test the models.” So far the Faughns have raised about a quarter of the $178,000 necessary to support Falk’s two-year proposal.
There is, of course, no guarantee.
“We don’t promise that we’re going to cure the disease,” Falk cautions. “But we think that this is the best chance to do it rationally and logically.”
“All of this is a race against time,” Dave says. “Katherine’s disease is progressive. We’ve had two MRIs, and if you look at them, you see the atrophy in the cerebellum. So the brain cells are dying. And when they die, there’s no bringing them back.”
Cali Spooner’s survival into her teens offers hope, but it is tenuous. “We don’t know where this disease is going to lead next,” Dave continues. “Mito diseases are generally multi-system diseases. Any part of your body that needs energy starts to be compromised. So you tend to see, over time, heart disease and liver disease and kidney disease. We don’t know what the future holds. So we’re desperate to find either a cure or a treatment that stops the progression as soon as we can, because we don’t know what tomorrow will bring for her.”
14q12-NM_025152.2(NUBPL)
When the family visited Philadelphia in October for Katherine to see a phalanx of specialists at CHOP, which now coordinates her care, Katherine showed a small child’s mastery of inhabiting the present day.
With the help of orthotic ankle braces, she now walks independently. While I talked with her parents, she scribbled letters and drawings on scraps of paper and pretended she was a mail carrier, delivering them around the table. She fell down half a dozen times, and got back up.
She told me about school: playing games with her friends Delaney and Ally, and counting dominoes for her math homework. Her articulation of words required substantial concentration to decipher, and occasionally Dave or Glenda would interject to make sure I was following. But one thing she said startled me, first by its clarity and later by its symbolic depth.
“What kinds of games do you like to play?” I asked her.
“Baby,” she said. “And family. And baby lion.”
“How do you play baby lion?” I asked.
“With a mommy lion and a daddy lion and a baby lion,” she said. Then, with a smile full of gusto, she expelled a sudden and fierce sound: a savage, exultant hissing growl.
She waited a beat to witness my reaction, and then said, quite clearly, “It’s a big roar for a little lion.”



Keep fighting!! God Bless!
This is our child and our exact journey…we even ended up with the same doctor at Cleveland Clinic. Our daughter is 22. She has had two five-month episodes of a (metabolic?) crisis, where her body odor changes so profoundly that I can smell it the moment I walk into the house even if she’s upstairs in her room. We Know this must be metabolic.
CHOP did her exome sequencing in summer 2015, but, unlike Katherine, we have not found any match. My daughter’s full case history can be found at https://docs.google.com/document/d/1PrbN8P68F_IqR0VQ2gDfhjqD71gpFhJ0Jwv11Y6W570/edit and we would appreciate any thoughts from other parents or researchers.
What a wonderful and extraordinary photograph on your cover! My thanks to the photographer, Jonathan Robert Willis. It crystallizes why we should continue to spend money on “orphan” diseases. The economics are real, but so is this wonderful little girl. I hope we can find a balance in fundamental research.
To the author of this article: You certainly have articulated just how complex and confounding rare diseases truly are, and the enormous frustrations, efforts and costs involved in protracted diagnosis and working against time…and the progression of the condition. However, I need to correct some medical information inherent in your piece. You state that Progeria is a result of a ACVR1 gene that turns soft tissue into bone. It is FOP (fibrodysplasia ossificans progressive) which does so, another ultra rare diseases, whose incidences are 1:2,000,000. The world wide expert, Dr. Fred Kaplan, resides in Penn’s distinguished orthopedic department. I have studied this disease myself for almost 20 years; I funded Dr. Kaplan”s medical chair as a tribute to my parents: The Isaac and Rose Nassau Professorship in Orthopedic Molecular Medicine in 1997. On the other hand, Progeria is another rare disease that causes extraordinarily accelerated aging in children, and is one of several porgeriod syndromes, resulting in small bodies, very old appearance, and a child who is 8 years old looking like he or she is 80. Because the Penn Gazelle is a well respected read and reference guide, it is important for those who read it and who might have a situation that requires further inquiry, that such information be properly represented and corrected. Otherwise, the wealth of information contained in this article is fascinating and profound. Diane Weiss